A lo largo de la historia, la medicina ha tenido que avanzar con una venda en los ojos. Durante milenios, nuestra profesión ha sido una ciencia de observación fortuita: veíamos un síntoma, probábamos una sustancia activa y esperábamos una respuesta en un esquema clásico de ensayo y error. Pero esa era de la "biología a ciegas" ha terminado oficialmente.
La llegada de AlphaFold 3 de Google DeepMind no representa una mejora tecnológica incremental; es un salto cuántico que transforma la práctica médica en una ingeniería molecular interactiva y predictiva.
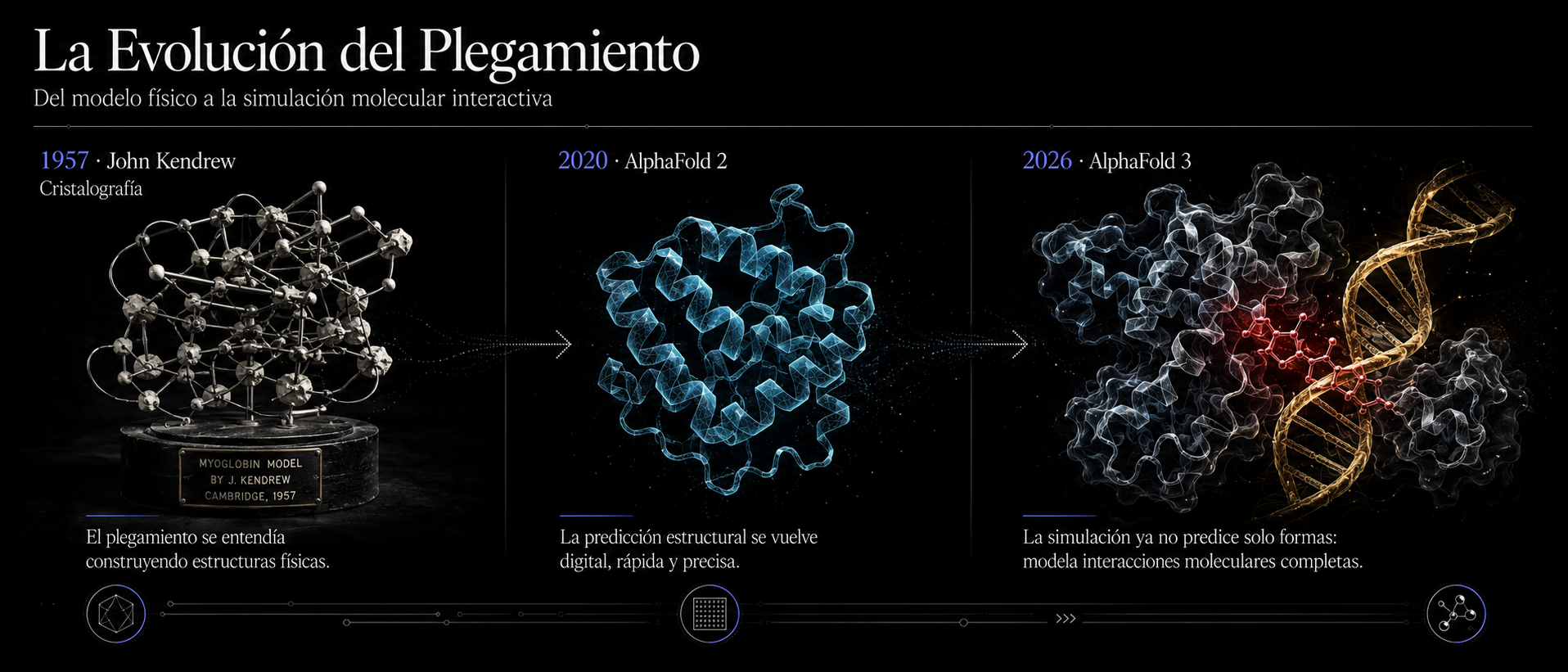
La transición histórica del plegamiento molecular: del primer modelo tridimensional físico de mioglobina en 1957 a la predicción digital en segundos de complejos atómicos enteros con AlphaFold 3.
Para dominar esta herramienta en el entorno clínico, hemos diseñado esta guía práctica dividida en cuatro entregas:
- Parte 1 (Este artículo): El cambio de paradigma biológico y la superación de los límites biofísicos tradicionales.
- Parte 2: La Anatomía del Algoritmo. Cómo funciona la inteligencia artificial por dentro a través de tokens, coevolución y difusión.
- Parte 3: Aplicación Clínica. El impacto directo de la medicina molecular digital en onco-inmunología, diseño de fármacos y longevidad.
- Parte 4: El Dilema de la Bioseguridad. Los riesgos de la tecnología dual y el debate ético sobre el acceso restringido a su código.
¿Qué significa esto para una clínica o especialista?
De acuerdo con el cambio de paradigma biológico expuesto, los profesionales de la salud pasan de observar el mapa genético a comprender la geometría tridimensional de la enfermedad en tiempo real. Esto permite superar los límites de la física tradicional, permitiendo que el conocimiento estructural molecular deje de ser un secreto de laboratorio y se convierta en una herramienta de medicina traslacional ágil e inmediata para evaluar interacciones celulares y fármacos.
El precio de la estructura: Carne de ballena y años de doctorado
Para valorar el impacto de AlphaFold 3, debemos recordar de dónde venimos. En los años 60, cuando el bioquímico John Kendrew intentó determinar por primera vez la estructura tridimensional de una proteína (la mioglobina), se enfrentá a un problema logístico masivo. Tras fracasar con corazones de caballo por la baja concentración de la molécula, tuvo que conseguir un gran trozo de carne de ballena de Perú para obtener cristales lo suficientemente grandes. Su trabajo requirió 12 años de esfuerzos minuciosos y le valió el Premio Nobel en 1962.
Hasta hace muy poco, las reglas básicas no habían cambiado mucho. Determinar la estructura tridimensional de una sola proteína mediante cristalografía de rayos X o criomicroscopía electrónica seguía costando decenas de miles de dólares y años de trabajo de laboratorio. Francamente, en el ámbito académico y de investigación médica, obtener la geometría de un par de proteínas solía ser el tema completo para la tesis de doctorado de un científico. La medicina avanzaba despacio porque la estructura molecular era un secreto celosamente guardado por la física.
La Paradoja de Levinthal: El reto computacional del millón de años
¿Por qué era tan difícil? Las proteínas nacen como una cadena lineal de aminoácidos, pero para funcionar como máquinas útiles dentro de la célula, deben plegarse sobre sí mismas adoptando una forma tridimensional sumamente compleja. Si se pliegan bien, la fisiología celular continúa; si se pliegan mal, nos enfrentamos a patologías como el Alzheimer, el Parkinson o el cáncer.
En 1969, el biólogo molecular Cyrus Levinthal realizó un cálculo estimado para ilustrar la complejidad de este proceso. Descubrió que incluso una proteína promedio pequeña puede adoptar una cantidad astronómica de configuraciones espaciales: 10³⁰⁰.

“La Paradoja de Levinthal demuestra que si una proteína promedio intentara plegarse probando de forma aleatoria todas sus combinaciones espaciales posibles, tardaría más que la edad del universo en encontrar su estado de menor energía. En la naturaleza, esto ocurre en milisegundos.”
Sin embargo, nuestras células resuelven este problema biofísico y pliegan miles de proteínas en una fracción de segundo.
La Singularidad de AlphaFold 3: De la foto estática a la coreografía celular
En 2020, AlphaFold 2 (cuyos creadores recibieron el Premio Nobel de Química en 2024) conmocionó a la ciencia al resolver este reto de cinco décadas. Sin embargo, la versión anterior tenía una limitación clínica importante: nos entregaba una "foto estática". Nos enseñaba la estructura tridimensional de una proteína aislada, sola en una habitación virtual vacía.
Pero como especialistas sabemos bien que la biología no ocurre en el aislamiento. La vida celular es una coreografía en movimiento, una danza caótica de interacciones complejas. Aquí es donde AlphaFold 3 cambia las reglas del juego de la medicina traslacional.
AlphaFold 2
Predice únicamente proteínas aisladas en estructuras estáticas limitadas al laboratorio.
AlphaFold 3
Predice complejos enteros interactuando: Proteína + ADN + ARN + Ligandos (fármacos) en tiempo real.
AlphaFold 3 ya no modela únicamente proteínas individuales. Ahora es capaz de predecir con precisión atómica el complejo entero en una sola plataforma: cómo una proteína se une al ADN para encender o apagar genes, cómo interactúa con el ARN y, lo más crítico para la práctica médica, cómo se acopla con los ligandos (fármacos y compuestos químicos).
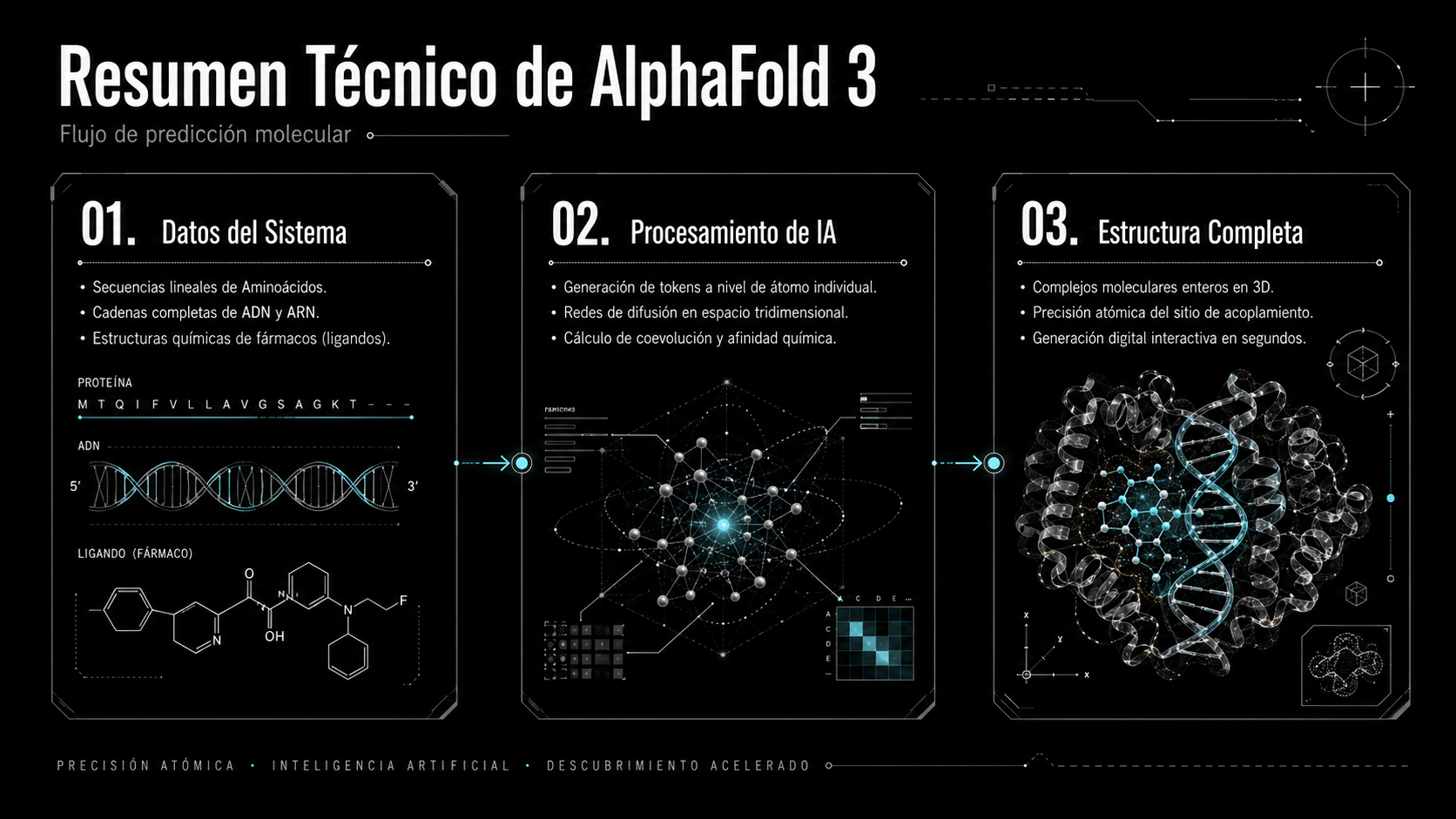
Síntesis estructural de AlphaFold 3: del código lineal del genoma y bases de datos a la visualización interactiva de interacciones químicas y proteínas en tiempo real.
Hemos pasado de observar el mapa genético a comprender la geometría tridimensional de la enfermedad en tiempo real. En la siguiente entrega, abriremos el capó de este software para entender, de forma visual y biofísica, la anatomía del algoritmo detrás de esta revolución.